

JMatPro® - Practical Software for Materials Properties
JMatPro® is a simulation software which calculates properties for alloys and is aimed at multi-component alloys used in industrial practice. It has been designed so that it can be used by any engineer or scientist that requires materials properties as part of their everyday work.

Our Products
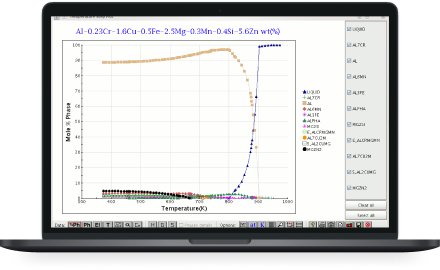
JMatPro® is a simulation software which calculates a wide range of materials properties for alloys and is particularly aimed at multi-component alloys used in industrial practice.
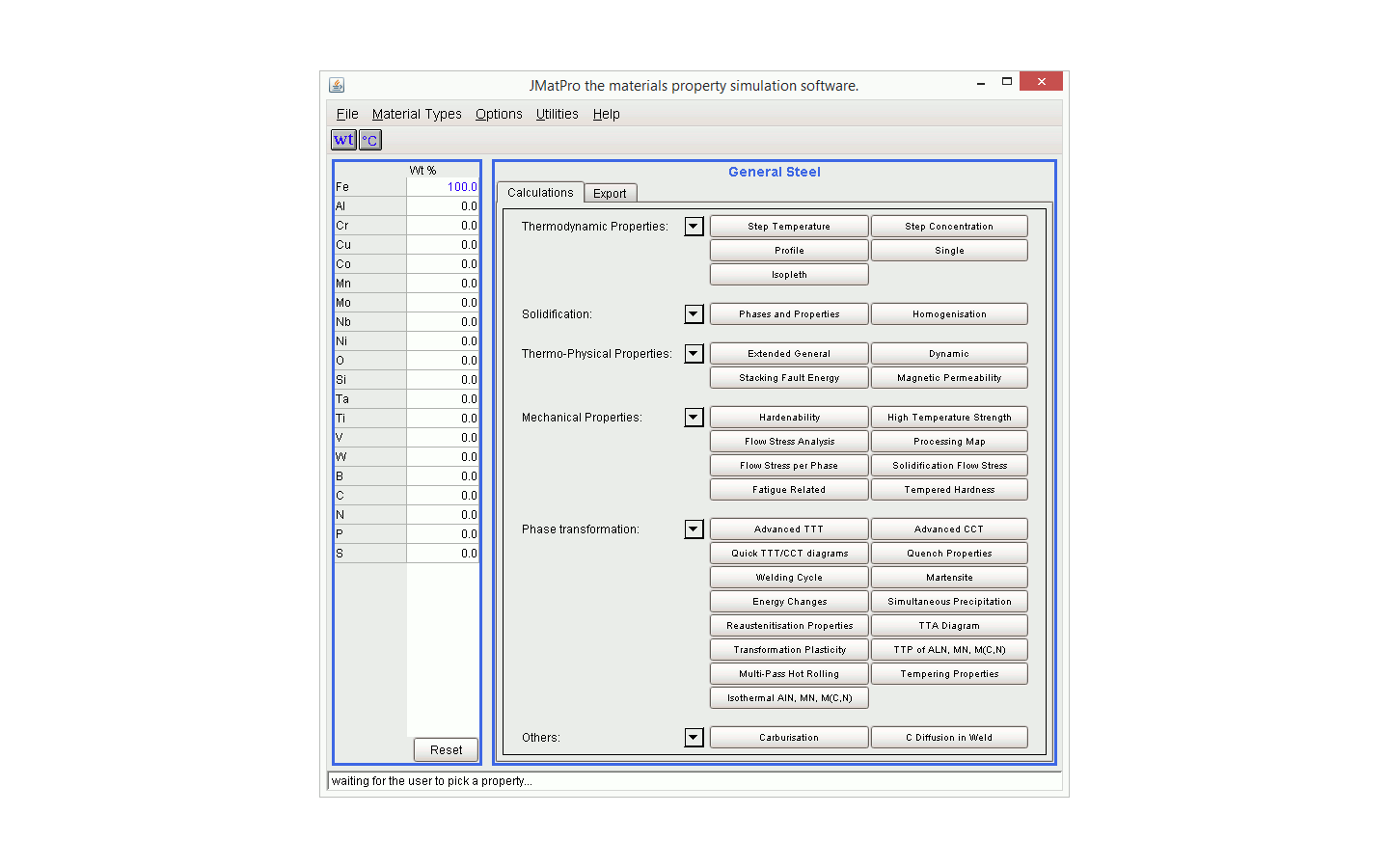
JMatPro®: The Core Product
You no longer need to scour handbooks or the literature resulting in a long and painful process with incomplete information. JMatPro® offers consistent and reliable modelling of these properties over a range of conditions all available from just one easy to use source.
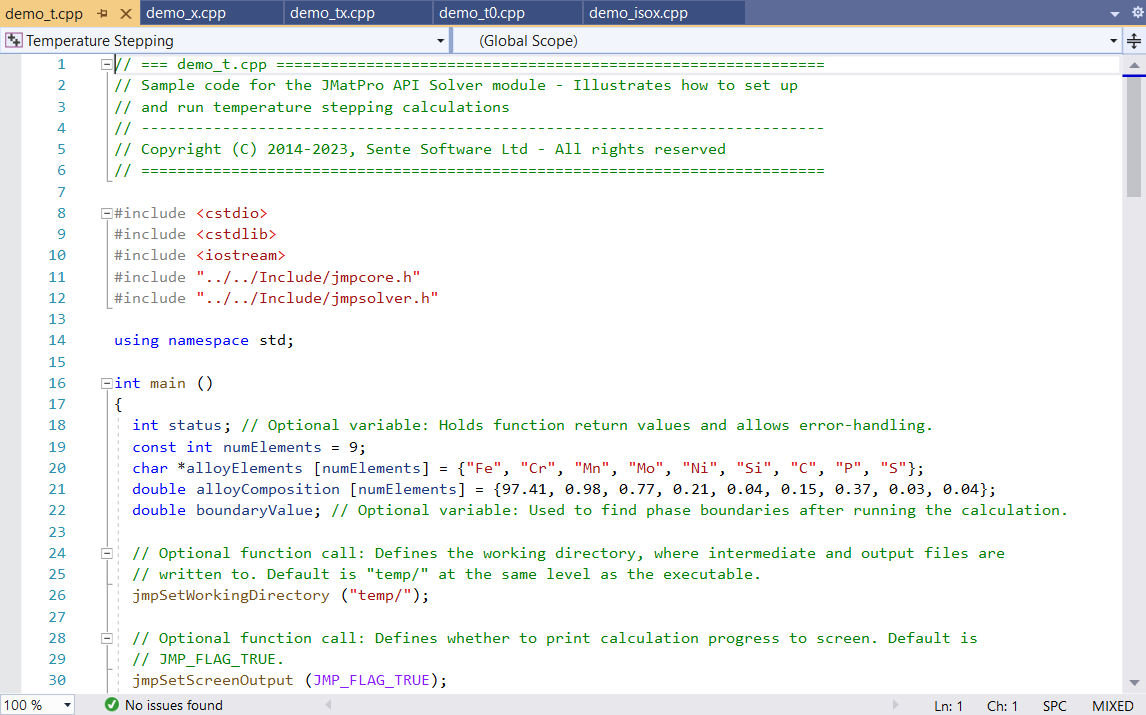
JMatPro® API
Incorporate specific functionality of the JMatPro® software into your own applications.
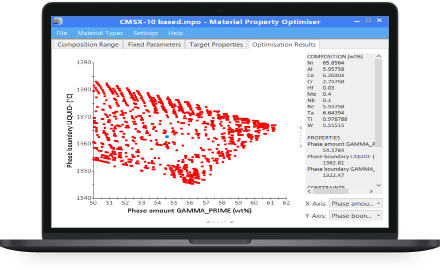
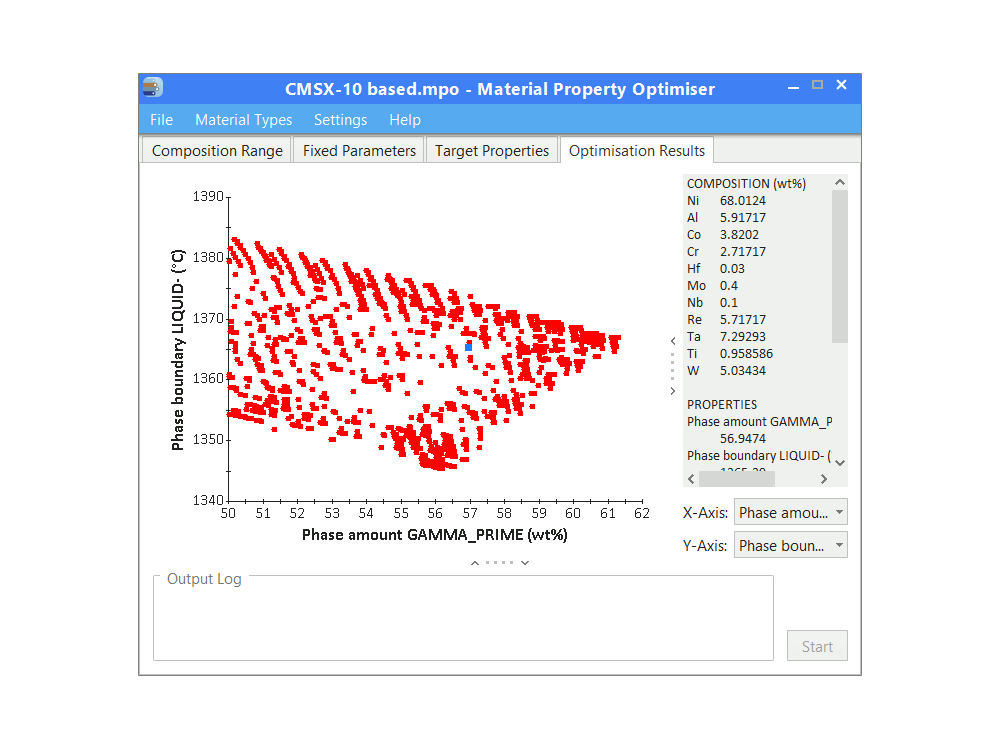
JMatPro® Material Property Optimiser (MPO)
A powerful optimisation tool for the design of multi-component alloys.

About Us
Sente Software Ltd. was created in 2001 to take responsibility for the long term commercial development of JMatPro®. It now leads the development of the new scientific capabilities in JMatPro® alongside the development of its powerful graphical user interface.
All our products combine industrial relevance with realistic physical models and user-friendly interfaces.
We have a proven track record for innovation and our products incorporate a thorough validation process.